
Lei Xiong
Postdoctoral Scholar
Genetics
Stanford University
Long-range cis-regulatory interactions (LRIs) spanning tens to hundreds of kilobases enable tissue-specific enhancers to coordinate complex gene expression programs essential for precise spatiotemporal control in development, cell differentiation, and disease. Systematically linking distal regulatory elements to their target genes remains a critical bottleneck limiting translation of GWAS discoveries into mechanistic insights and therapeutic strategies. The central goal of this proposal is to develop interpretable deep learning models that decode long-range regulatory syntax and predict the causal impact of distal noncoding elements and variants on chromatin state and gene expression. Using CAD (Coronary Artery Disease) macrophages as a model system, I will develop and test a generalizable framework for identifying long-range regulatory circuits and understand their role in CAD. Central Hypothesis: Long-range gene regulation is governed by a learnable sequence syntax, and disruption of this syntax by distal noncoding variants contributes to complex disease through dysregulation of enhancer-promoter communication.
Feel free to reach out by email jsxlei at gmail.com or other social media listed above!
Education
Tsinghua University
Ph.D. in Computational Biology
Advisor: Prof.Qiangfeng Cliff Zhang
Thesis: Artificial intelligence method for single-cell ATAC-seq data via feature extraction
University of Science and Technology of China (USTC)
B.S. in Biology, Shitsan Pai Talent Program in Life Sciences
Advisor: Prof.Nieng Yan
Thesis: Structure basis and transport mechanism of membrane protein GLUT3
News
Released chrombpnet_pytorch for reproducing the official chrombpnet in pytorch
Our work Deep learning prediction of ribosome profiling with Translatomer reveals translational regulation and interprets disease variants was published on Nature Machine Intelligence.
Our work A versatile informative diffusion model for single-cell ATAC-seq data generation and analysis was accepted by NeurIPS 2024
Awarded School of Medicine Dean’s Postdoctoral Fellowship, Stanford.
Join Prof.Anshul Kundaje lab in Genetics at Stanford, also co-supervised by Prof.Mike Bassik.
Our workshop paper scCLIP: Multi-modal Single-cell Contrastive Learning Integration Pre-training was presented on NeurIPS 2023 AI4Science workshop
Our work Online single-cell data integration through projecting heterogeneous datasets into a common cell-embedding space was published on Nature Communications.
Join Prof.Manolis Kellis lab in CSAIL at MIT and Broad Institute of MIT and Harvard.
Awarded Outstanding Doctoral Dissertation Award of Tsinghua University.
Awarded Outstanding Graduate of Beijing.
PhD. thesis Defense.
Our work SCALE was awarded Top 10 Algorithms and Tools for Bioinformatics in China in 2019.
Our work SCALE was awarded Top 10 Advances in Bioinformatics in China in 2019
Our work SCALE method for single-cell ATAC-seq analysis via latent feature extraction was published on Nature Communications.
Publications
2025

Single-cell multiregion epigenomic rewiring in Alzheimer’s disease progression and cognitive resilience.
Cell 2025.Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by progressive cognitive decline, yet its epigenetic underpinnings remain elusive. Here, we generate and integrate single-cell epigenomic and transcriptomic profiles of 3.5 million cells from 384 postmortem brain samples across 6 regions in 111 AD and control individuals. We identify over 1 million candidate cis-regulatory elements (cCREs), organized into 123 regulatory modules across 67 cell subtypes. We define large-scale epigenomic compartments and single-cell epigenomic information and delineate their dynamics in AD, revealing widespread epigenome relaxation and brain-region-specific and cell-type-specific epigenomic erosion signatures during AD progression. These epigenomic stability dynamics are closely associated with cell-type proportion changes, glial cell-state transitions, and coordinated epigenomic and transcriptomic dysregulation linked to AD pathology, cognitive impairment, and cognitive resilience. This study provides critical insights into AD progression and cognitive resilience, presenting a comprehensive single-cell multiomic atlas to advance the understanding of AD.
Cancer cell phagocytosis induces an anti-inflammatory gene regulatory program in macrophages.
bioRxiv 2025.Macrophages are capable of eliminating cancer cells by phagocytosis, particularly in the presence of monoclonal antibody (mAb) therapies targeting tumor antigens. Paradoxically, tumor-associated macrophages are typically associated with poor patient outcome, and can promote tumor growth by secretion of immunosuppressive cytokines and growth factors. The mechanisms by which these pro-tumor macrophage states arise are poorly understood, and it is unclear how mAb-induced cancer cell phagocytosis may contribute to these states. To understand how antibody-dependent cancer cell phagocytosis (ADCP) alters macrophage state and function, we profiled gene expression and chromatin accessibility changes over time after ADCP. We observed that after ADCP, macrophages upregulate an anti-inflammatory gene regulatory program, characterized by expression of pro-angiogenic and immunosuppressive chemokine genes, and increased activity by cellular, oxidative, and lysosomal stress transcription factors. This gene regulatory program was shared among phagocytic macrophages following either ADCP or apoptotic cancer cell phagocytosis, in addition to substrate-specific pathways. Conditioned media from macrophages promoted EMT in cancer cells, but this pro-EMT macrophage phenotype was attenuated following ADCP, but not following apoptotic cancer cell phagocytosis. The phagocytic gene signature we identified in vitro is also expressed by tumor-associated macrophages across numerous cancer types in vivo . Together, this work identifies an anti-inflammatory and immunosuppressive epigenetic program in macrophages following ADCP upon mAb treatment, and expands our understanding of how phagocytosis influences macrophage heterogeneity in the tumor microenvironment.
2024
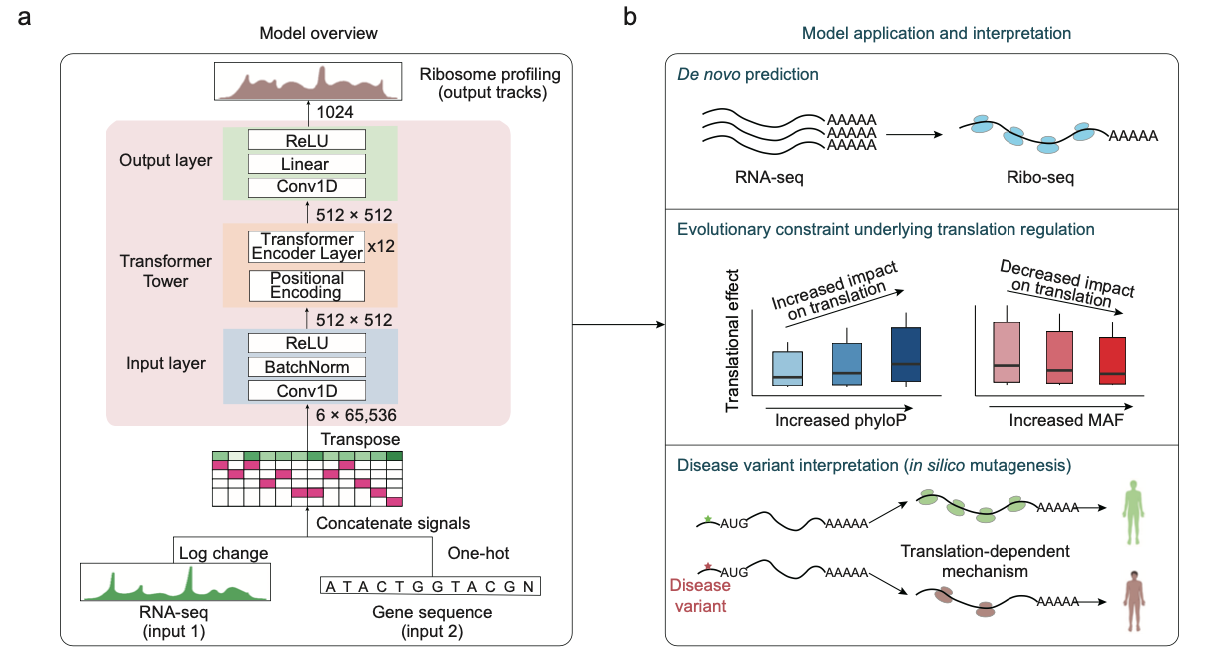
Deep learning prediction of ribosome profiling with Translatomer reveals translational regulation and interprets disease variants.
Nat. Mach. Intell. 2024.Gene expression involves transcription and translation. Despite large datasets and increasingly powerful methods devoted to calculating genetic variants’ effects on transcription, discrepancy between messenger RNA and protein levels hinders the systematic interpretation of the regulatory effects of disease-associated variants. Accurate models of the sequence determinants of translation are needed to close this gap and to interpret disease-associated variants that act on translation. Here we present Translatomer, a multimodal transformer framework that predicts cell-type-specific translation from messenger RNA expression and gene sequence. We train the Translatomer on 33 tissues and cell lines, and show that the inclusion of sequence improves the prediction of ribosome profiling signal, indicating that the Translatomer captures sequence-dependent translational regulatory information. The Translatomer achieves accuracies of 0.72 to 0.80 for the de novo prediction of cell-type-specific ribosome profiling. We develop an in silico mutagenesis tool to estimate mutational effects on translation and demonstrate that variants associated with translation regulation are evolutionarily constrained, both in the human population and across species. In particular, we identify cell-type-specific translational regulatory mechanisms independent of the expression quantitative trait loci for 3,041 non-coding and synonymous variants associated with complex diseases, including Alzheimer’s disease, schizophrenia and congenital heart disease. The Translatomer accurately models the genetic underpinnings of translation, bridging the gap between messenger RNA and protein levels as well as providing valuable mechanistic insights for uninterpreted disease variants.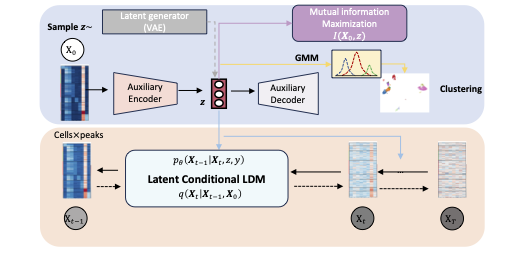
A versatile informative diffusion model for single-cell ATAC-seq data generation and analysis.
NeurIPS 2024.The rapid advancement of single-cell ATAC sequencing (scATAC-seq) technologies holds great promise for investigating the heterogeneity of epigenetic landscapes at the cellular level. The amplification process in scATAC-seq experiments often introduces noise due to dropout events, which results in extreme sparsity that hinders accurate analysis. Consequently, there is a significant demand for the generation of high-quality scATAC-seq data in silico. Furthermore, current methodologies are typically task-specific, lacking a versatile framework capable of handling multiple tasks within a single model. In this work, we propose ATAC-Diff, a versatile framework, which is based on a latent diffusion model conditioned on the latent auxiliary variables to adapt for various tasks. ATAC-Diff is the first diffusion model for the scATAC-seq data generation and analysis, composed of auxiliary modules encoding the latent high-level variables to enable the model to learn the semantic information to sample high-quality data. Gaussian Mixture Model (GMM) as the latent prior and auxiliary decoder, the yield variables reserve the refined genomic information beneficial for downstream analyses. Another innovation is the incorporation of mutual information between observed and hidden variables as a regularization term to prevent the model from decoupling from latent variables. Through extensive experiments, we demonstrate that ATAC-Diff achieves high performance in both generation and analysis tasks, outperforming state-of-the-art models.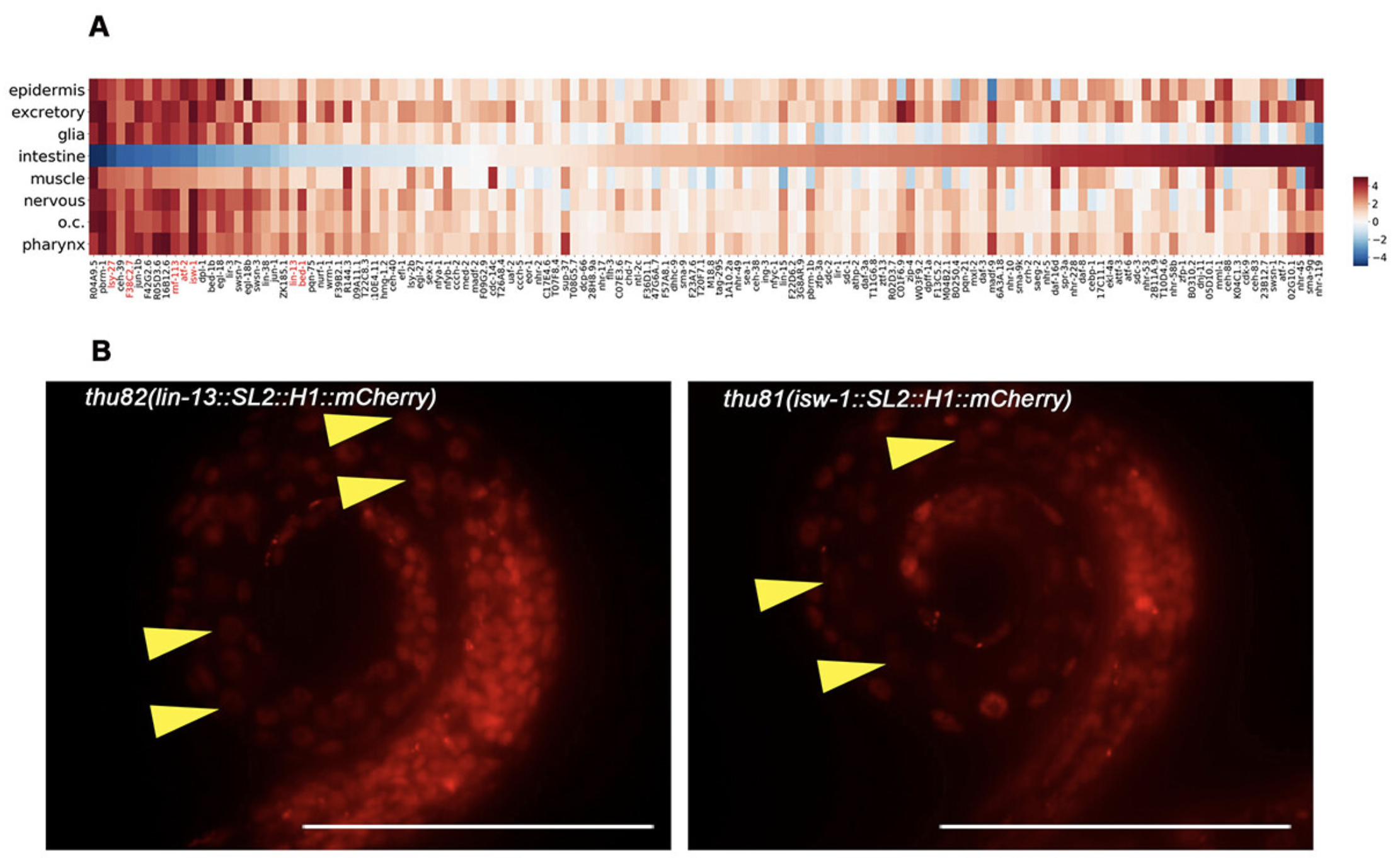
Tissue-specific silencing of integrated transgenes achieved through endogenous RNA interference in Caenorhabditis elegans.
RNA Biol. 2024.Transgene silencing is a common phenomenon observed in Caenorhabditis elegans, particularly in the germline, but the precise mechanisms underlying this process remain elusive. Through an analysis of the transcription factors profile of C. elegans, we discovered that the expression of several transgenic reporter lines exhibited tissue-specific silencing, specifically in the intestine of C. elegans. Notably, this silencing could be reversed in mutants defective in endogenous RNA interference (RNAi). Further investigation using knock-in strains revealed that these intestine-silent genes were indeed expressed in vivo, indicating that the organism itself regulates the intestine-specific silencing. This tissue-specific silencing appears to be mediated through the endo-RNAi pathway, with the main factors of this pathway, mut-2 and mut-16, are significantly enriched in the intestine. Additionally, histone modification factors, such as met-2, are involved in this silencing mechanism. Given the crucial role of the intestine in reproduction alongside the germline, the transgene silencing observed in the intestine reflects the self-protective mechanisms employed by the organisms. In summary, our study proposed that compared to other tissues, the transgenic silencing of intestine is specifically regulated by the endo-RNAi pathway.
2023
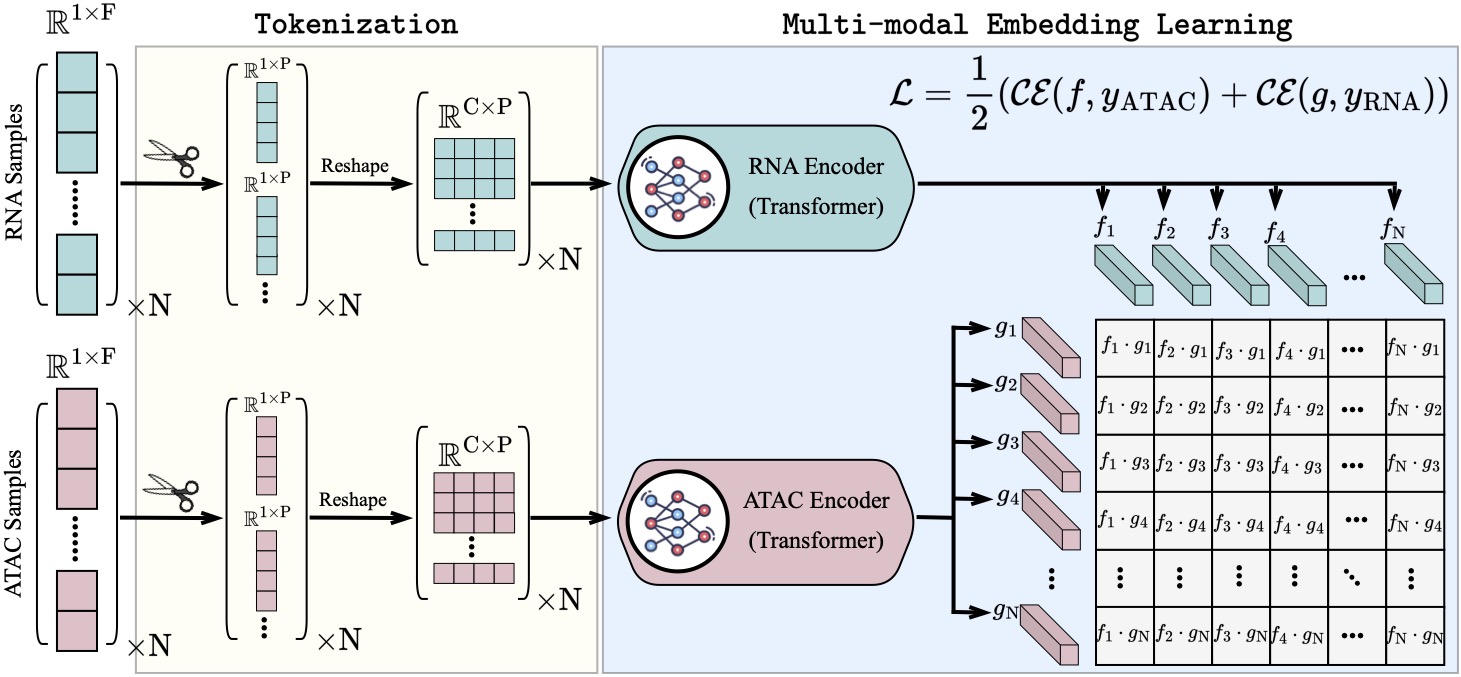
scCLIP: Multi-modal Single-cell Contrastive Learning Integration Pre-training.
NeurIPS AI for Science workshop 2023.Recent advances in multi-modal single-cell sequencing technologies enable the simultaneous profiling of chromatin accessibility and transcriptome in individual cells. Integration analysis of multi-modal single-cell data offers a more comprehensive understanding of the regulatory mechanisms linking chromatin status and gene expression, driving cellular processes and diseases. In order to acquire features that align peaks and genes within the same embedding space and facilitate seamless zero-shot transfer to new data, we introduced scCLIP (single-cell Contrastive Learning Integration Pretraining), a generalized multi-modal transformer model with contrastive learning. We show that this model outperforms other competing methods, and beyond this, scCLIP learns transferable features across modalities and generalizes to unseen datasets, which pose the great potential to bridge the vast number of unpaired unimodal datasets both existing and new data generated in the future. Specifically, we propose the first large-scale transformer model designed for single-cell ATAC-seq data by patching peaks across the genomes and representing each patch as a token. This innovative approach enables us effectively to address the scalability challenges posed by scATAC-seq, even when dealing with datasets of up to one million dimensions. Codes are provided at: https://github.com/jsxlei/scCLIP.
2022
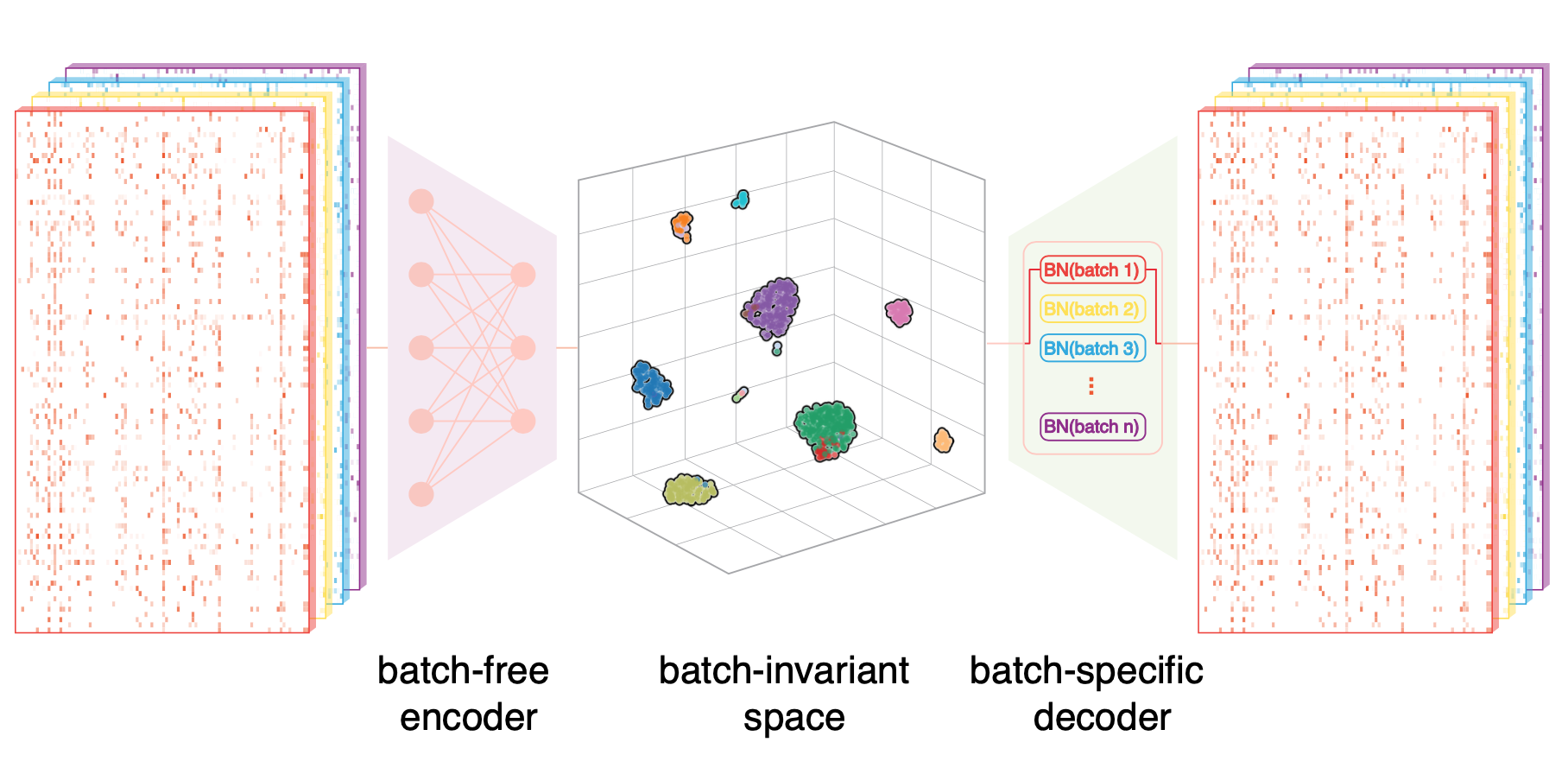
Online single-cell data integration through projecting heterogeneous datasets into a common cell-embedding space.
Nat. Commun. 2022.Computational tools for integrative analyses of diverse single-cell experiments are facing formidable new challenges including dramatic increases in data scale, sample heterogeneity, and the need to informatively cross-reference new data with foundational datasets. Here, we present SCALEX, a deep-learning method that integrates single-cell data by projecting cells into a batch-invariant, common cell-embedding space in a truly online manner (i.e., without retraining the model). SCALEX substantially outperforms online iNMF and other state-of-the-art non-online integration methods on benchmark single-cell datasets of diverse modalities, (e.g., single-cell RNA sequencing, scRNA-seq, single-cell assay for transposase-accessible chromatin use sequencing, scATAC-seq), especially for datasets with partial overlaps, accurately aligning similar cell populations while retaining true biological differences. We showcase SCALEX’s advantages by constructing continuously expandable single-cell atlases for human, mouse, and COVID-19 patients, each assembled from diverse data sources and growing with every new data. The online data integration capacity and superior performance makes SCALEX particularly appropriate for large-scale single-cell applications to build upon previous scientific insights.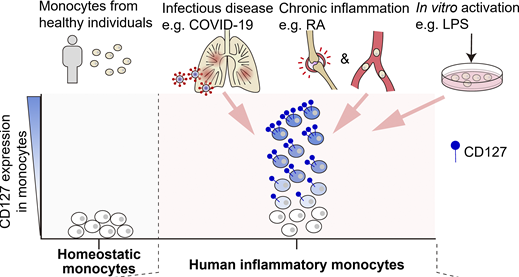
CD127 imprints functional heterogeneity to diversify monocyte responses in inflammatory diseases.
J. Exp. Med. 2022.Inflammatory monocytes are key mediators of acute and chronic inflammation; yet, their functional diversity remains obscure. Single-cell transcriptome analyses of human inflammatory monocytes from COVID-19 and rheumatoid arthritis patients revealed a subset of cells positive for CD127, an IL-7 receptor subunit, and such positivity rendered otherwise inert monocytes responsive to IL-7. Active IL-7 signaling engaged epigenetically coupled, STAT5-coordinated transcriptional programs to restrain inflammatory gene expression, resulting in inverse correlation between CD127 expression and inflammatory phenotypes in a seemingly homogeneous monocyte population. In COVID-19 and rheumatoid arthritis, CD127 marked a subset of monocytes/macrophages that retained hypoinflammatory phenotypes within the highly inflammatory tissue environments. Furthermore, generation of an integrated expression atlas revealed unified features of human inflammatory monocytes across different diseases and different tissues, exemplified by those of the CD127high subset. Overall, we phenotypically and molecularly characterized CD127-imprinted functional heterogeneity of human inflammatory monocytes with direct relevance for inflammatory diseases.
2019
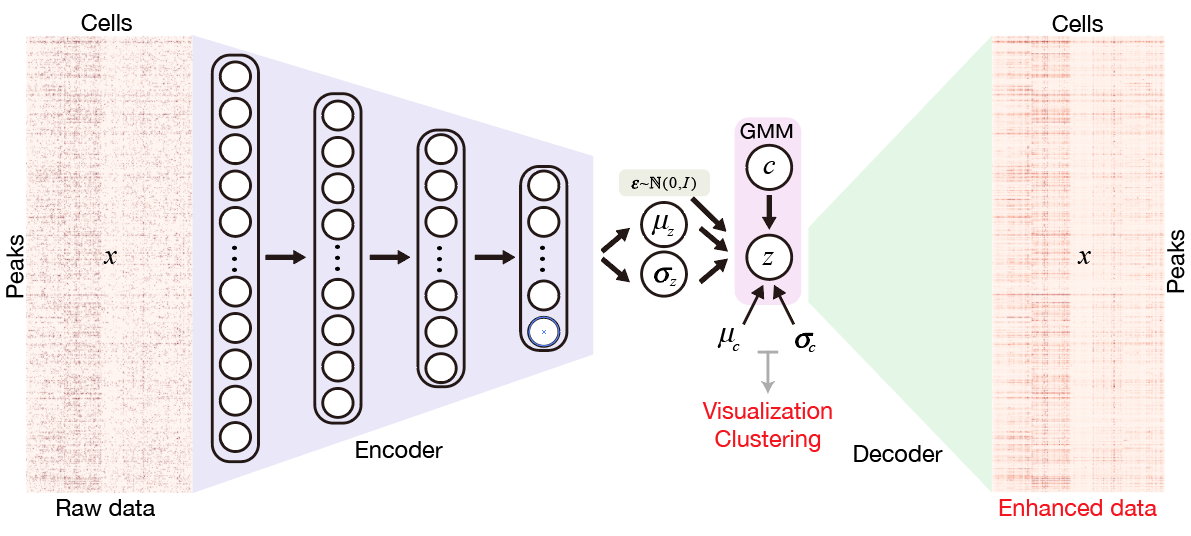
SCALE method for single-cell ATAC-seq analysis via latent feature extraction.
Nat. Commun. 2019.Single-cell ATAC-seq (scATAC-seq) profiles the chromatin accessibility landscape at single cell level, thus revealing cell-to-cell variability in gene regulation. However, the high dimensionality and sparsity of scATAC-seq data often complicate the analysis. Here, we introduce a method for analyzing scATAC-seq data, called Single-Cell ATAC-seq analysis via Latent feature Extraction (SCALE). SCALE combines a deep generative framework and a probabilistic Gaussian Mixture Model to learn latent features that accurately characterize scATAC-seq data. We validate SCALE on datasets generated on different platforms with different protocols, and having different overall data qualities. SCALE substantially outperforms the other tools in all aspects of scATAC-seq data analysis, including visualization, clustering, and denoising and imputation. Importantly, SCALE also generates interpretable features that directly link to cell populations, and can potentially reveal batch effects in scATAC-seq experiments.
2015
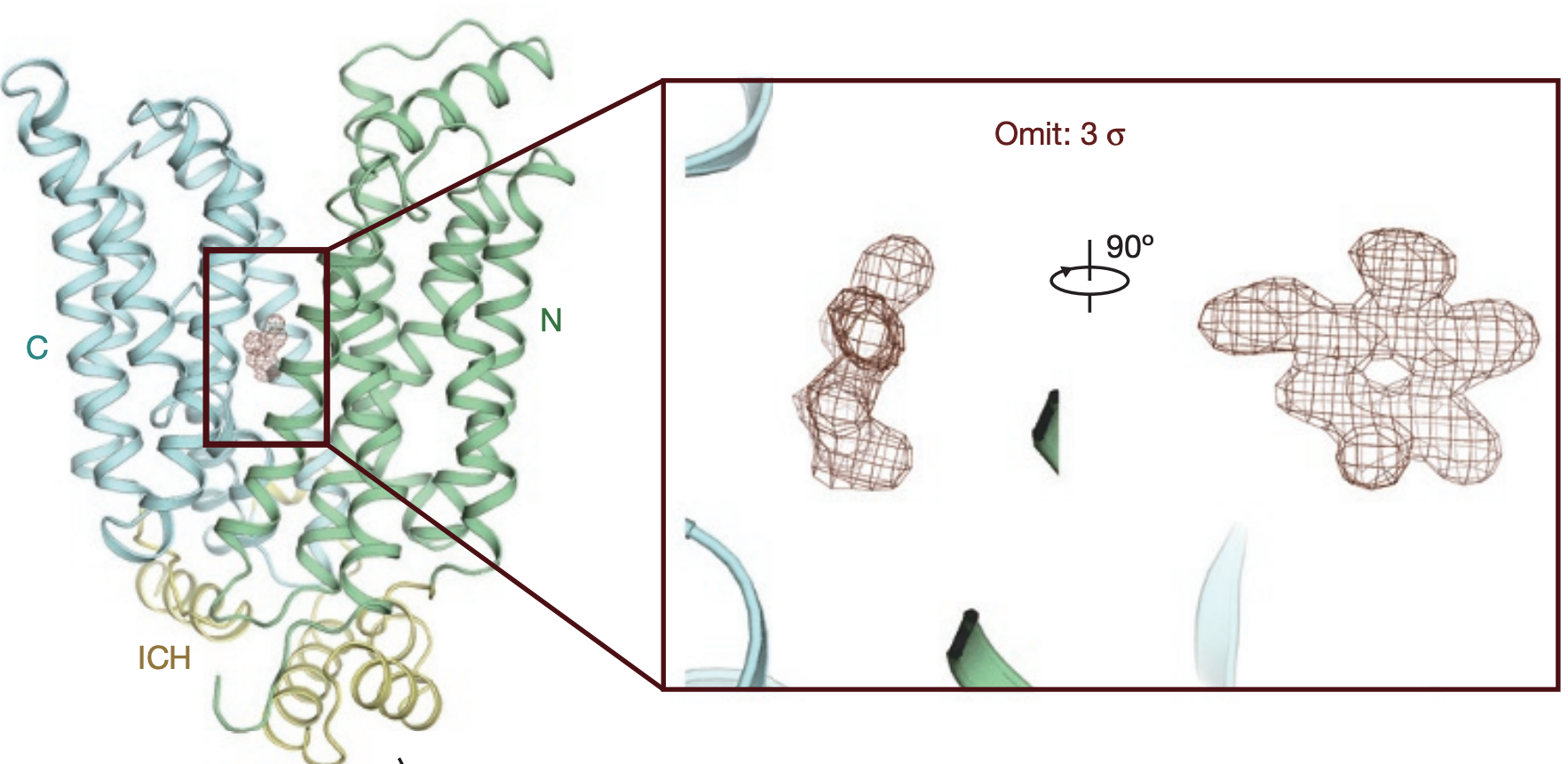
Molecular basis of ligand recognition and transport by glucose transporters.
Nature 2015.The major facilitator superfamily glucose transporters, exemplified by human GLUT1-4, have been central to the study of solute transport. Using lipidic cubic phase crystallization and microfocus X-ray diffraction, we determined the structure of human GLUT3 in complex with D-glucose at 1.5 Å resolution in an outward-occluded conformation. The high-resolution structure allows discrimination of both α- and β-anomers of D-glucose. Two additional structures of GLUT3 bound to the exofacial inhibitor maltose were obtained at 2.6 Å in the outward-open and 2.4 Å in the outward-occluded states. In all three structures, the ligands are predominantly coordinated by polar residues from the carboxy terminal domain. Conformational transition from outward-open to outward-occluded entails a prominent local rearrangement of the extracellular part of transmembrane segment TM7. Comparison of the outward-facing GLUT3 structures with the inward-open GLUT1 provides insights into the alternating access cycle for GLUTs, whereby the C-terminal domain provides the primary substrate-binding site and the amino-terminal domain undergoes rigid-body rotation with respect to the C-terminal domain. Our studies provide an important framework for the mechanistic and kinetic understanding of GLUTs and shed light on structure-guided ligand design.
Softwares










Honors and Awards
School of Medicine Dean’s Postdoctoral Fellowship, Stanford.
Outstanding Doctoral Dissertation of Tsinghua University (Top 5%).
Outstanding Graduate of Beijing (Top 5%).
Top 10 Advances in Bioinformatics in China in 2019.
Top 10 Algorithms and Tools for Bioinformatics in China in 2019.
Outstanding Fellowship, Advanced Innovation Center of Structure Biology, Tsinghua.
Innovation Fellowship, Advanced Innovation Center of Structural Biology, Tsinghua.
iGEM Gold Medal, USTC-China.
2012-2013 Student Scholarship, USTC.
2011-2012 Student Scholarship, USTC.
Freshman Scholarship, USTC.